Definition
Arachnoiditis literally means "inflammation of the arachnoid," which is the middle of the three membranes (meninges) surrounding the brain and spinal cord. The term more generally refers to several rare neurologic disorders caused by inflammation of a portion of the arachnoid and subarachnoid space, affecting the neural tissue that lies beneath. Symptoms of arachnoiditis are quite variable, and may include anything from a skin rash to moderate or severe pain, to paralysis. The condition is often progressive, can only rarely be cured, and existing treatments vary in their effectiveness.
Description
Three membranes, including the dura mater, arachnoid, and pia mater, and a layer of cerebrospinal fluid (CSF) surround, protect, and cushion the brain and spinal cord. The pia mater adheres to the brain and spinal cord, and is separated from the arachnoid membrane by the subarachnoid space, which contains the circulating CSF. Arachnoiditis always involves inflammation in one or several restricted areas, but the entire membrane is never affected. Fibrous (scar) tissue growth along the affected section of the membrane usually occurs, projecting down through the subarachnoid space and encompassing neural tissue of the brain (cerebral arachnoiditis) and/or nerve roots of the spinal cord (spinal arachnoiditis). Nerve damage occurs through restricted blood flow (ischemia), compression from accumulated fluids (edema), and secondary effects of the inflammatory process itself.
Other terms used less frequently for arachnoiditis include arachnitis, chronic adhesive arachnoiditis (CAA), and spinal fibrosis. Other conditions that may be associated with or mimic arachnoiditis include syringomyelia (cyst near the spinal cord), cauda equina (lower spinal cord) syndrome, and spinal tumor. Several different types of arachnoiditis have been described, including adhesive (fibrous attachments), ossifying (bony tissue growth), neo-plastic (tumor growth), optochiasmatic (optic nerve and chiasm), and rhinosinusogenic (olfactory nerve and area above the sinuses).
Demographics
The true incidence of arachnoiditis is not known, but it is rare. It affects males and females equally, and seems to be less frequent in children than in adults. Rare cases of familial arachnoiditis have been documented, but no particular ethnic groups seem to be at higher risk.
Causes and symptoms
The causes of arachnoiditis are varied, but fall into the following four categories:
* trauma to the membrane due to spinal surgery (often multiple procedures), cranial or spinal injury, or needle insertion to remove CSF for testing
* external agents such as anesthesia, corticosteroids, medications, or medical dyes/chemicals injected near the spinal cord (epidural) or directly into the CSF
* infection of the arachnoid/CSF (meningitis)
* blood in the CSF caused by trauma, spontaneous bleeding, or infection
For reasons that are not entirely clear, different areas of the arachnoid have differing sensitivities to the causative agents. Spinal arachnoiditis due to infection most often occurs in the cervicothoracic (neck and upper back) region, while cases due to external agents most often occur in the lumbosacral (lower back) area. Likewise, spinal arachnoiditis of any type is more common than the cerebral/cranial variety.
Symptoms of cerebral arachnoiditis may include severe headaches, vision disturbances, dizziness, and nausea/vomiting. Vision disturbances are especially pronounced in optochiasmatic arachnoiditis. If inflammation and tissue growth in specific areas of the cranial arachnoid membrane divert or obstruct normal flow of the CSF, the result is hydrocephalus (increased fluid pressure within the brain).
Typical symptoms of spinal arachnoiditis include back pain that increases with activity, pain in one or both legs or feet, and sensory abnormalities of some type, usually involving decreased reflexes. Patients may also exhibit decreased range of motion of the trunk or legs, and urinary sphincter dysfunction (urgency, frequency, or incontinence). In more severe cases, partial or complete paralysis of the lower extremities may occur.
Diagnosis
The most reliable method of establishing the diagnosis of arachnoiditis is a positive computed tomography (CT) or magnetic resonance imaging (MRI) scan, combined with one or more of the symptoms. Testing for certain cell types and proteins in the CSF may prove helpful only in the early stages of the inflammation. On the other hand, imaging studies may be negative or equivocal early on, and only later be more definitive as inflammation and tissue growth becomes more pronounced. In some cases, a definitive diagnosis may not be possible.
Treatment team
A neurologist is the primary specialist involved in monitoring and treating arachnoiditis. Occupational/physical therapy (OT/PT) might also be suggested to assist with treatment for pain and adaptation to sensory deficits and/or muscular weakness in the back and lower limbs. A neurosurgeon performs any elected surgeries to address the various effects of the inflammation. Many individuals with chronic pain attend pain clinics staffed by physicians (usually anesthesiologists) and nurses who specialize in pain management. Neuropsychiatrists and neuropsychologists specialize in treating the psychological problems specific to individuals who have an underlying neurologic condition.
Treatment
Treatment for arachnoiditis is mostly done with medications, and is geared toward reducing the inflammation and alleviating pain. Medications may include both nonsteroidal and steroidal anti-inflammatory drugs, along with non-narcotic and narcotic pain medications. Other possible treatments include epidural steroid injections, transcutaneous electrical nerve stimulation (TENS), topical analgesics, and alternative medical therapies.
Direct spinal cord stimulation is a newer pain management method that involves placement of tiny electrodes under the skin, directly on the affected nerve roots near the spine. Mild current application inhibits pain signals, and is provided by a small, battery-powered unit that is placed under the skin by a surgeon.
Surgery to remove fibrous or ossified tissue at the site of the inflammation is used only if more conservative methods do not provide sufficient relief. Surgical removal of a small portion of one or more vertebrae at the area of the nerve root is called a laminectomy. A neurosurgeon treats hydrocephalus by placing a shunt (plastic tube) from the brain to the abdominal cavity to relieve increased pressure. Microsurgical techniques to remove scar tissue from around the nerve roots themselves are a more recent development.
Prognosis
Given the lack of effective treatments for arachnoiditis, the prognosis in most instances is poor, with the neurologic symptoms remaining static or worsening over time. It is not uncommon for people who undergo surgery for the condition to improve at first, but eventually regress within several years.
Resources
BOOKS
Bradley, Walter G., et al., eds. Neurology in Clinical Practice, 3rd ed. Boston: Butterworth-Heinemann, 2000.
Victor, Maurice, and Allan H. Ropper. Adam's and Victor's Principles of Neurology, 7th ed. New York: The McGraw-Hill Companies, Inc., 2001.
Wiederholt, Wigbert C. Neurology for Non-Neurologists, 4th ed. Philadelphia: W.B. Saunders Company, 2000.
PERIODICALS
Chin, Cynthia T. "Spine Imaging." Seminars in Neurology 22 (June 2002): 205–220.
Faure, Alexis, et al. "Arachnoiditis Ossificans of the Cauda Equina: Case Report and Review of the Literature." Journal of Neurosurgey/Spine 97 (September 2002): 239–243.
Rice, M. Y. K., et al. "Obstetric Epidurals and Chronic Adhesive Arachnoiditis." British Journal of Anaesthesia 92 (2004): 109–120.
Wright, Michael H., and Leann C. Denney "A Comprehensive Review of Spinal Arachnoiditis." Orthopaedic Nursing 22 (May/June 2003): 215–219.
ORGANIZATIONS
American Paraplegia Society. 75-20 Astoria Boulevard, Jackson Heights, NY 11370-1177. (718) 803-3782.
American Syringomyelia Alliance Project, Inc. P.O. Box 1586, Longview, TX 75606-1586. 800-272-7282.
NIH/NINDS Brain Resources and Information Network. PO Box 5801, Bethesda, MD 20824. (800) 352-9424.
National Organization for Rare Disorders (NORD). 55 Kenosia Ave, PO Box 1968, Danbury, CT 06813-1968. (800) 999-6673; Fax: (203) 798-2291.
National Spinal Cord Injury Association. 6701 Democracy, Bethesda, MD 20817. (800) 962-9629.
Spinal Cord Society. 19051 County Hwy 1, Fergus Falls, MN 56537. (218) 739-5252.
Arachnoiditis
Autonomic dysfunction
Definition
Dysfunction of the autonomic nervous system (ANS) is known as dysautonomia. The autonomic nervous system regulates unconscious body functions, including heart rate, blood pressure, temperature regulation, gastrointestinal secretion, and metabolic and endocrine responses to stress such as the "fight or flight" syndrome. As regulating these functions involves various and multiple organ systems, dysfunctions of the autonomic nervous systems encompass various and multiple disorders.
Description
The autonomic nervous system consists of three subsystems: the sympathetic nervous system, the parasympathetic nervous system and the enteric nervous system. The ANS regulates the activities of cardiac muscle, smooth muscle, endocrine glands, and exocrine glands. The autonomic nervous system functions involuntarily (reflexively) in an automatic manner without conscious control.
In contrast to the somatic nervous system that always acts to excite muscles groups, the autonomic nervous systems can act to excite or inhibit innervated tissue. The ANS achieves this ability to excite or inhibit activity via a dual innervation of target tissues and organs. Most target organs and tissues are innervated by neural fibers from both the parasympathetic and sympathetic systems. The systems can act to stimulate organs and tissues in opposite ways (antagonistic). For example, parasympathetic stimulation acts to decrease heart rate. In contrast, sympathetic stimulation results in increased heart rate. The systems can also act in concert to stimulate activity. The autonomic nervous system achieves this control via two divisions: the sympathetic nervous system and the parasympathetic nervous system. Dysfunctions of the autonomic nervous system are recognized by the symptoms that result from failure of the sympathetic or parasympathetic components of the ANS.
Primary dysautonomias include multiple system atrophy (MSA) and familial dysautonomia. The dysfunction can be extensive and manifest as a general autonomic failure or can be confined to a more localized reflex dysfunction.
With multiple system atrophy, a generalized autonomic failure, male patients experience urinary retention or incontinence and impotence (an inability to achieve or maintain a penile erection). Both males and females experience ataxia (lack of muscle coordination) and a dramatic decline in blood pressure when they attempt to stand (orthostatic hypotension). Symptoms similar to Parkinson's disease may develop, such as slow movement, tremors, and stiff muscles. Visual disturbances, sleep disturbances, and decreased sweating may also occur.
Persons with autonomic dysfunction who do not exhibit the classical symptoms of orthostatic hypotension may exhibit a less dramatic dysfunction termed orthostatic intolerance. These patients experience a milder fall in blood pressure when attempting to stand. However, because the patients have an increased heart rate when standing, they are described as having postural tachycardia syndrome (POTS).
Although not as prevalent in the general population as hypertension, orthostatic intolerance is the second most common disorder of blood pressure regulation and is the most prevalent autonomic dysfunction. Orthostatic hypotension and orthostatic intolerance can result in a wide array of disabilities. Common orthostatic intolerance syndromes include: hyperadrenergic orthostatic hypotension (partial dysautonomia); orthostatic tachycardia syndrome (sympathicotonic orthostatic hypotension); postural orthostatic tachycardia syndrome (mitral valve prolapse syndrome); postural tachycardia syndrome (soldier's heart); hyperadrenergic postural hypotension (vasoregulatory asthenia); sympathotonic orthostatic hypotension (neurocirculatory asthenia); hyperdynamic beta-adrenergic state (irritable heart syndrome); and idiopathic hypovolemia (orthostatic anemia).
Demographics
Milder forms of autonomic dysfunction such as orthostatic intolerance affect an estimated 500,000 people in the United States. Orthostatic intolerance more frequently affects women; female-to-male ratio is at least 4:1. It is most common in people less than 35 years of age. More severe forms of dysautonomia such as multiple system atrophy often occur later in life (average age of onset 60 years) and affect men four times as often as women.
Causes and symptoms
Symptoms of the autonomic dysfunction of orthostatic intolerance include lightheadedness, palpitations, weakness, and tremors when attempting to assume an upright posture. Less frequently, patients experience visual disturbances, throbbing headaches, and often complain of fatigue and poor concentration. Some patients report fainting when attempting to stand.
The cause of lightheadedness, fainting, and similar symptoms is a lack of adequate blood pressure in the cerebral circulatory system.
In addition to orthostatic hypotension and Parkinson-type symptoms, persons with multiple systems atrophy may have difficulty articulating speech, sleep apnea and snoring, pain in the back of the neck, and fatigue. Eventually, cognitive (mental reasoning) ability declines in about 20% of cases. Multiple systems atrophy occurs sporadically and the cause is unknown.
Diagnosis
Diagnosis of orthostatic intolerance is made when a patient experiences a decrease of blood pressure (not exceeding 20/10 mm Hg) when attempting to stand and a heart rate increase of less than 30 beats per minute.
Diagnosis of other types of dysautonomia is difficult, as the disorders are varied and mimic other diseases of the nervous system. As Parkinsonism is the most frequent motor deficit seen in multiple systems atrophy, it is often misdiagnosed as Parkinson's disease. Magnetic resonance imaging (MRI) of the brain can sometimes detect abnormalities of striatum, cerebellum, and brainstem associated with multiple systems atrophy. But in up to 20% of MSA patients, MRI of the brain is normal. A test with the drug clonidine has also been used to differentiate Parkinson's disease from multiple systems atrophy, as certain hormone levels in the blood will increase in persons with Parkinson's disease after clonidine administration, but not in persons with multiple systems atrophy. Symptoms such as severe dysarthria (difficulty articulating speech) and stridor (noisy inspiration) alert the physician to the possibility of multiple systems atrophy, as they occur in the disorder, but are rare in Parkinson's disease.
Treatment team
Caring for a person with a disorder of the autonomic nervous system requires a network of health professionals, community resources, and friends or family members. A neurologist usually makes the diagnosis, and the neurologist and primary physician coordinate ongoing treatment and symptom relief. Physical, occupational, speech, and respiratory therapists provide specialized care, as do nurses. Social service and mental health consultants organize support services.
Treatment
At present there is no cure for severe autonomic dysfunction. Treatment is centered on the remediation of symptoms, patient support, and the treatment of underlying diseases and disorders in cases of secondary autonomic dysfunction. In many cases, cure or an improvement in the underlying disease or disorder improves the patient prognosis with regard to remediation of autonomic dysfunction symptoms.
With regard to orthostatic hypotension, drug treatment includes fludrocortisone, ephedrine, or midodrine. Medications are accompanied by postural relief such as elevation of the bed at the head and by dietary modifications to provide some relief for the symptoms of dizziness and tunnel vision.
In multiple systems atrophy, anti-Parkinson medications such as Sinemet often help with some of the symptoms of muscle rigidity and tremor, and create an overall feeling of well-being. Medications used in the treatment of orthostatic hypotension tend to not perform as well in this group; although they elevate the blood pressure while standing, they decrease the blood pressure while reclining.
Recovery and rehabilitation
Recovery from some dysautonomias can be complicated by secondary conditions such as alcoholism, diabetes, or Parkinson's disease. Some conditions improve with treatment of the underlying disease, while only halting of the progression of symptoms is accomplished in others. Some mild dysautonomias stabilize and, with treatment, cause few limitations to daily activities.
Overall, as there are no cures for most severe or progressive dysautonomias, the emphasis is instead placed upon maintaining mobility and function for as long as possible. Aids for walking and reaching, positioning devices, and strategies for maintaining posture, balance, and blood pressure while rising can be provided by physical and occupational therapists. Speech and nutritional therapists can devise diets and safe strategies for eating, and recommend tube feedings if necessary.
Clinical trials
As of mid-2004, the Mount Sinai Medical Center in New York was recruiting participants for a study related to a new drug for the treatment of multiple systems atrophy. Persons interested in participating in the study (Droxidopa in Treating Patients With Neurogenic Hypotension) should contact the study recruiting coordinator Horacio Kaufmann at telephone: (212) 241-7315. Additional trials for the study and treatment of multiple systems atrophy and other dysautonomias can be found at the National Institutes of Health website for clinical trials:
Prognosis
The prognosis for persons suffering autonomic dysfunction is variable and depends on specific dysfunction and on the severity of the dysfunction. Autonomic dysfunctions can present as acute and reversible syndromes, or can present in more chronic and progressive forms. Persons with orthostatic intolerance can usually maintain a normal lifespan and active lifestyle with treatment and minimal coping measures, while persons with multiple systems atrophy usually have a lifespan of about 5–7 years after diagnosis.
BOOKS
Goldstein, David S., and Linda J. Smith. The NDRF Handbook for Patients with Dysautonomias. Malden, MA: Blackwell Futura Media, 2002.
OTHER
"Disorders of the Autonomic Nervous System." National Dysautonomia Research Foundation. May 16, 2004 (May 22, 2004).
"NINDS Dysautonomia Information Page." National Institute of Neurological Disorders and Stroke. May 16, 2004 (May 22, 2004).
ORGANIZATIONS
Dysautonomia Foundation. 633 Third Avenue, 12th Floor, New York, NY 10017-6706. (212) 949-6644; Fax: (212) 682-7625. info@familialdysautonomia.org.
Familial Dysautonomia Hope Foundation, Inc. (FD Hope). 1170 Green Knolls Drive, Buffalo Grove, IL 60089. (828) 466-1678. info@fdhope.org.
National Dysautonomia Research Foundation. 1407 West 4th Street, Red Wing, MN 55066-2108. (651) 267-0525; Fax: (651) 267-0524. ndrf@ndrf.org.
National Organization for Rare Disorders (NORD). P.O. Box 1968 (55 Kenosia Avenue), Danbury, CT 06813-1968. (203) 744-0100 or (800) 999-NORD; Fax: (203) 798-2291. orphan@rarediseases.org.
Shy-Drager/Multiple System Atrophy Support Group, Inc. 2004 Howard Lane, Austin, TX 78728. (866) 737-4999 or (800) 999-NORD; Fax: (512) 251-3315. Don.Summers@shy-drager.com.
Acute disseminated encephalomyelitis
Definition
Acute disseminated encephalomyelitis (ADE) is a neurological disorder involving inflammation of the brain and spinal cord. A hallmark of the disorder is damage to the myelin sheath that surrounds the nerve fibers in the brain, which results in the inflammation.
Description
Acute disseminating encephalomyelitis was first described in the mid-eighteenth century. The English physician who first described the disorder noted its association with people who had recently recovered from smallpox. Symptoms often develop without warning. As well, mental disorientation can occur. The disorder is also known as postinfectious encephalomyelitis and immune-mediated encephalomyelitis. The nerve demyelination that occurs in ADE also occurs in multiple sclerosis. However, the two maladies differ in that multiple sclerosis is long lasting and can recur over time, while ADE has a monophasic course, meaning that once it is over, further attacks rarely occur.
Demographics
ADE can occur in both children and adults, although it occurs more commonly in children. ADE is not rare, accounting for approximately 30% of all cases of encephalitis (brain inflammation).
Causes and symptoms
Acute disseminating encephalomyelitis can occur as a consequence of a bacterial or viral infection (including HIV), following recovery from infection with the malarial protozoan, or as a side effect of vaccination or another inoculation. ADE is usually a consequence of a viral illness, and occurs most often after measles, followed by rubella, chicken pox, Epstein-Barr, mumps and pertussis (whooping cough). Typically, symptoms appear two to three weeks after the precipitating infection or immunization. Alternatively, ADE may develop with no known associations.
Despite the different causes, the symptoms that develop are similar. A number of non-specific symptoms, which vary from one person to another, include headache, stiff neck, fever, vomiting, and weight loss. These symptoms are quickly followed by lethargic behavior, seizures, hallucinations, sight difficulties, and even coma. Paralysis can occur in an arm or leg (monoparesis) or along an entire side of the body (hemiplegia).
These symptoms can last a few weeks to a month. In some people, symptoms can progress from the appearance of symptoms to coma and death in only a few days. Brain damage is largely confined to the white matter. Microscopic examination will typically reveal invasion of white blood cells into small veins. The nerve myelin damage occurs in the regions where the white blood cells accumulate. Examination of the brains of patients who have died of the disorder has not yielded consistent results. Some brains appear normal, while others display the nerve damage and white blood cell congestion.
Diagnosis
Diagnosis is made based on the above symptoms and the patient's medical history (i.e., recent infection or vaccination). In the early stages of the disorder, diagnosis can be confused with diseases including acute meningitis, acute viral encephalitis, and multiple sclerosis. Often, the latter can be ruled out using magnetic resonance imaging (MRI) and examination of the cerebrospinal fluid (CSF). Typically, in acute disseminating encephalomyelitis, CSF contains abnormally elevated levels of white blood cells and protein; and magnetic resonance imaging can reveal brain alterations.
Treatment team
The treatment team typically consists of a primary care physician and, when hospitalization is necessary, nurses and specialized medical care personnel.
Treatment
Corticosteroid medication is often prescribed in order to lessen the nerve inflammation. Use of high doses of steroids can often produce a rapid diminishing of the symptoms. Other kinds of treatment depend on the nature of the symptoms that develop. Supportive care includes keeping a patient comfortable and hydrated.
Recovery and rehabilitation
Persons recovering from acute disseminated encephalomyelitis need time to recover their normal consciousness and movements. Problems with memory, especially short-term memory, may be present. The recovering person sometimes has trouble controlling their emotions and is easily frustrated. Frequent periods of rest, alternating with shorter periods of mental and physical exercise are prescribed during initial recovery. The maximum possible recovery of brain and motor function may take a period of weeks or months.
Clinical trials
There are no clinical trials for the study of ADE recruiting patients or being planned in the United States, as of January 2004. However, organizations such as the National Institute for Neurological Disorders and Stroke undertake and fund studies on disorders that involve damage to the myelin sheath of nerve cells. By understanding the nature of the disorders, it is hoped that detection can be improved and strategies will evolve to prevent or reverse the nerve damage.
Prognosis
Prognosis varies from person to person. Some patients may recover fully, with no residual effects. Others may have some residual damage. Seldomly, ADE is fatal. Early detection and treatment improves a patient's outlook.
Special concerns
Although the incidence of ADE occurring after vaccination is rare, in recent years, public debate has led some parents to choose that their children not receive the recommended childhood vaccinations. The American Academy of Pediatrics asserts that, despite concerns about vaccine safety, vaccination is far safer than accepting the risks for the diseases that the vaccines prevent.
Resources
BOOKS
Icon Health Publications. The Official Patient's Sourcebook on Acute Disseminated Encephalomyelitis: A Revised and Updated Directory for the Internet Age. San Diego: Icon Group International, 2002.
PERIODICALS
Anlar, B., C. Basaran, G. Kose, A. Guven, S. Haspolat, A. Yakut, A. Serdaroglu, N. Senbil, H. Tan, E. Karaagaoglu, and K. Oguz. "Acute disseminated encephalomyelitis in children: outcome and prognosis." Neuropediatrics (August 2003): 194–199.
Brass, S. D., Z. Caramanos, C. Santos, M. E. Dilenge, Y. Lapierre, and B. Rosenblatt. "Multiple sclerosis vs acute disseminated encephalomyelitis in childhood." Pediatric Neurology (September 2003): 227–231.
Koibuchi, T., T. Nakamura, T. Miura, T. Endo, H. Nakamura, T. Takahashi, H. S. Kim, Y. Wataya, K. Washizaki, K. Yoshikawa, and A. Iwamoto. "Acute disseminated encephalomyelitis following Plasmodium vivax malaria." Journal of Infection and Chemotherapy (September 2003): 254–256.
Narciso, P., S. Galgani, B. Del Grosso, M. De Marco, A. De Santis, P. Balestra, V. Ciapparoni, and V. Tozzi. "Acute disseminated encephalomyelitis as manifestation of primary HIV infection." Neurology (November 2001): 1493–1496.
OTHER
"Acute Disseminated Encephalomyelitis Information Page." National Institute of Neurological Disorders and Stroke.
ORGANIZATIONS
National Institute for Neurological Diseases and Stroke (NINDS). 6001 Executive Boulevard, Bethesda, MD 20892. (301) 496-5751 or (800) 352-9424.
National Organization for Rare Disorders. 55 Kenosia Avenue, Danbury, CT 06813-1968. (203) 744-0100 or (800) 999-6673; Fax: (203) 798-2291.
Brian Douglas Hoyle, PhD
Postoperative Pain in Neurosurgery: A Pilot Study in Brain Surgery.
Clinical Studies
Neurosurgery. 38(3):466-470, March 1996.De Benedittis, Giuseppe M.D.; Lorenzetti, Ariberto M.D.; Migliore, Matteo M.D.; Spagnoli, Diego M.D.; Tiberio, Francesca M.D.; Villani, Roberto M. M.D.
Abstract:
THE INCIDENCE, MAGNITUDE, and duration of acute pain experienced by neurosurgical patients after various brain operations are not precisely known, because of a lack of well-designed clinical and epidemiological studies. We assessed these important pain variables in 37 consecutive patients who underwent various brain neurosurgical procedures. Postoperative pain was more common than generally assumed (60%). In two-thirds of the patients with postoperative pain, the intensity was moderate to severe. Pain most frequently occurred within the first 48 hours after surgery, but a significant number of patients endured pain for longer periods. Pain was predominantly superficial(86%), suggesting somatic rather than visceral origin and possibly involving pericranial muscles and soft tissues. Subtemporal and suboccipital surgical routes yielded the highest incidence of postoperative pain. Age and sex were significantly associated with the onset of pain, with female and younger patients reporting higher percentages of postoperative pain. Psychological Minnesota Multiphasic Personality Inventory profiles of patients with and without pain significantly differed on the Hypochondriasis scale, with patients without pain scoring unexpectedly higher than patients with pain. It is possible that hypochondriasis serves as a defense mechanism against pain, at least in some patients. Results of this pilot study indicate that postoperative pain after brain surgery is an important, although neglected, clinical problem, that deserves greater attention by surgical teams, to provide better and more appropriate treatment.
Copyright (C) by the Congress of Neurological Surgeons
Endoscope-assisted Brain Surgery: Basic Concept, and Current Technique.
Special Technical Article
Neurosurgery. 42(2):219-224, February 1998.Perneczky, Axel MD; Fries, Georg MD
Abstract:
RATIONALE: The evolution of neurosurgical techniques indicates the effort to reduce surgery-related traumatization of patients. The reduction of traumatization contributes to better postoperative outcomes. The improvement of diagnostic imaging techniques facilitates not only the precise localization of lesions but also the accurate determination of topographical relations of specific lesions to individual anatomic variations of intracranial structures. This precision of diagnostic imaging should be used to perform individual surgical procedures through so-called keyhole approaches. Keyhole craniotomies are afflicted with a reduction of light intensity in the depth of the operating field, and they provide rather narrow viewing angles. Thus, objects located directly opposite the approach entrance are more visible than those in the shadow of the microscope beam. These two deficiencies of keyhole craniotomies can be compensated for by the intraoperative use of rigid rod lens endoscopes, the shaft of which remains easily controllable through the surgical microscope.
CONCEPT: Endoscope-assisted microsurgery, like all routine microsurgical procedures, is performed with both hands; the endoscope is fixed in its desired position via a mechanical arm to the headholder. Because of their superior optical quality and maneuverability, only rigid lens scopes are used for endoscope-assisted brain microsurgery. There are five ways of observing the endoscopic and microscopic images at the same time: 1) observation of the microscopic image through the oculars of the microscope and observation of the endoscopic image on a video screen placed in front of the surgeon, 2) observation of the microscopic image through the oculars of the microscope and display of the endoscopic image on a head-mounted LCD screen, 3) projection of both microscopic and endoscopic images on one screen in a picture-in-picture mode, 4) projection of both microscopic and endoscopic images into specially designed microscope oculars, and 5) transmission of both microscopic and endoscopic images into a head-mounted LCD screen.
DISCUSSION: With the knowledge of almost all individual anatomic and pathoanatomic details of a specific patient, it is possible to target the individual lesion through a keyhole approach using the particular anatomic windows. As the light intensity and the depiction of important anatomic details are improved by the intraoperative use of lens scopes, endoscope-assisted microsurgery during keyhole approaches may provide maximum efficiency to remove the lesion, maximum safety for the patient, and minimum invasiveness.
Clinical Studies
Neurosurgery. 42(2):226-231, February 1998.Fries, Georg MD; Perneczky, Axel MD
Abstract:
OBJECTIVES: Microsurgical techniques and instruments that help to reduce intraoperative retraction of normal intracranial neuronal and vascular structures contribute to improved postoperative results. To achieve sufficient control of the operating field without retraction of neurovascular components, the resection of dura and bone edges is frequently required, which, on the other hand, increases operating time and operation-related trauma. The use of endoscopes may help to reduce retraction and, at the same time, may help to avoid additional dura and bone resection. The aim of this study is to describe the principles on which the technique of endoscope-assisted brain surgery is based, to give an impression of possible indications for endoscope-assisted microsurgical procedures, and, with illustrative cases, to delineate the advantages of endoscopes used as surgical instruments during microsurgical approaches to intracranial lesions.
METHODS: During a period of 4.5 years, 380 microsurgical procedures were performed as endoscope-assisted microneurosurgical operations. This surgical series was analyzed for time of surgery, usefulness of intraoperative endoscopy, and complication rates. Lens scopes with viewing angles of 0 to 110 degrees and with diameters of 2.0 to 5.0 mm as well as newly designed "viewing dissectors" (curved, rigid fiberscopes) with diameters of 1.0 to 1.5 mm connected to a video unit were used as microsurgical instruments. The positioning of the endoscopes was achieved by retractor arms fixed to the Mayfield headholder. Thus, the surgeon was able to perform customary microsurgical manipulations with both hands under simultaneous endoscopic and microscopic control.
RESULTS: The lesions treated with endoscope-assisted microsurgery comprised 205 tumors, 53 aneurysms, 86 cysts, and 36 neurovascular compression syndromes. Eighty-nine of these lesions were localized in the ventricular system, 242 in the subarachnoid space or intracerebral, and 49 in the sella. Endoscope-assisted microsurgery was advantageous to reduce the size and the operation-related tissue trauma of approaches to lesions within the ventricular system, in the brain tissue as well as in the subarachnoid space at the base of the brain. Using less retraction during tumor removal, the visual control of retrosellar, endosellar, retroclival, and infratentorial structures was improved. Video-endoscope instrumentation was especially helpful during procedures in the posterior cranial fossa and at the craniocervical junction. It allowed for inspection of channels and hidden structures (e.g., the internal auditory meatus, the ventral surface of the brain stem, the ventral aspect of root entry zones of cranial nerves, the content of the foramen magnum, and the upper cervical canal), both without retraction and without resection of dura and bone edges. Endoscope instrumentation during surgery for large or giant aneurysms was useful to dissect perforators on the back side of the aneurysms and to control the completeness of clipping.
CONCLUSION: Although the results reported herein cannot be compared directly with those of exclusive microsurgical procedures performed during the same period of time, videoendoscope-assisted microsurgery can be recommended as a time-saving, trauma-reducing procedure apt to improve postoperative outcomes.
Copyright (C) by the Congress of Neurological Surgeons
Ventriculomegaly, isolated
Gianluigi Pilu, MD
Synonyms: ventriculomegaly, hydrocephalus, acqueductal stenosis, communicating hydrocephalus
Definition: overt enlargement of the lateral ventricles (atrial width > 15 mm) in the absence of other sonographically demonstrable central nervous system anomalies.
Prevalence: The incidence of congenital cerebral lateral ventriculomegaly ranges between 0.3 to 1.5 in 1000 births in different series.Isolated ventriculomegaly accounts for 30%-60% of fetuses with enlarged lateral cerebral ventricles.
Pathogenesis: In the majority of cases, isolated cerebral lateral ventriculomegaly is the consequence of an obstruction along the normal pathway of the cerebrospinal fluid (obstructive hydrocephalus).
Etiology: Congenital ventriculomegaly is a heterogeneous disease for which genetic, infectious, teratogenic and neoplastic causes have been implicated. A multifactorial pattern of inheritance is probably responsible for most cases of congenital hydrocephalus. X-linked hydrocephalus comprises approximately 5% of all cases. This condition is caused by mutations in the gene at Xq28 encoding for L1, a neural cell adhesion molecule. Mutations in this gene are also responsible for other syndromes with clinical overlap that are frequently referred to as the X-linked hydrocephalus spectrum and include MASA (mental retardation, aphasia, shuffling gait, adducted thumbs), complicated X-linked spastic paraplegia (SP 1), X-linked mental retardation-clasped thumb (MR-CT) syndrome, and some forms of X-linked agenesis of the corpus callosum. Infections implicated in the determination of congenital ventriculomegaly include toxoplasmosis, syphilis, cytomegalovirus, mumps and influenza virus.
Pathology: Overt lateral ventriculomegaly can result from different pathological entities. Fetuses with isolated ventriculomegaly diagnosed in utero at our institution were usually found at birth to have either aqueductal stenosis or communicating ventriculomegaly. Progression from communicating ventriculomegaly to aqueductal stenosis has been documented and it is uncertain whether these two conditions are separate clinical entities. Communicating hydrocephalus may however derive from acute events such as subarachnoid hemorrhage, or be caused by overproduction of cerebro-spinal fluid by a choroid plexus papilloma. The degree of ventricular enlargement is variable. Knowledge about the pathogenesis of congenital ventriculomegaly is largely incomplete. Thinning of the cortex, macrocrania and symptoms of intracranial hypertension are frequently found. Studies performed in experimental animals and based on biopsies of brain tissue obtained in children at the time of shunting seem to demonstrate the following sequence of events: initially there is disruption of the ependymal lining, followed by edema of the white matter. This phase has been considered reversible. Later, there is proliferation of astrocytes and fibrosis of the white matter. The gray matter seems to be spared during the initial staged of the process.
Recurrence risk: apart from X-linked hydrocephalus (recurrence risk 50% of males) congenital ventriculomegaly is mostly multifactorial. Couples with a previously affected child have a recurrence risk of 4%.
Associated anomalies: extra-cranial abnormalities occur in 30% of cases. Chromosomal aberrations are found in 11% of cases (6% of fetuses with ventriculomegaly as the only antenatal finding, 25% of cases with multiple anomalies). The X-linked hydrocephalus spectrum is frequently associated with abduction of the thumbs, abnormal facies and absence of the septum pellucidum.
Diagnosis: Different approaches have been proposed for the diagnosis of fetal lateral cerebral ventriculomegaly. A qualitative approach has been suggested. Under normal condition, the large fetal choroid plexus entirely fills the cavity of the lateral ventricle at the level of the atria, being closely apposed to both the medial and the lateral wall, irrespective of gestational age. In even early stages of ventriculomegaly, the choroid plexus is shrunken anteriorly displaced, thus being clearly detached from the medial wall (Figure 1). However, in a finite number of normal fetuses some disproportion between the choroid plexus and the atrial lumen is found. Normal values for virtually all portions of the lateral ventricles throughout gestation are available. Measurement of the width of atrium is however favored. Between 15 and 40 weeks’ gestation, the mean value is consistently about 7 mm and the standard deviation about 1 mm in most studies. Some degree of asymmetry of the lateral ventricles exists in human fetal brain and is detectable in utero.] A measurement of less than 10 mm is indicative of normalcy. Overt lateral cerebral ventriculomegaly is defined as a measurement of 15 mm or more. Sonographic demonstration of abducted thumbs in combination with ventriculomegaly and other intracranial abnormalities should prompt the diagnosis of X-linked hydrocephalus spectrum.
Figure 1: Fetal hydrocephalus: gross enlargement of the lateral ventricles, thinning of the cortex, asymmetric choroid plexuses.
Differential diagnosis: The main problem is distinguishing isolated ventriculomegaly from more complex abnormalities of the fetal brain that have frequently a different prognosis (e.g. holoprosencephaly, porencephaly, etc). We recommend careful multiplanar examination of the fetal brain, performed if possible with a high-resolution vaginal probe, and a detailed evaluation of the spine.
Implications for targeted examinations: For patients at risk for fetal cerebral ventriculomegaly (e.g. because of a previously affected child, or because of TORCH infection), we recommend multiplanar examination of the fetal brain, performed if possible with a high-resolution vaginal probe, including visualization and assessment of both lateral ventricle. It has been our experience, and it has been reported in a handful of cases that ventriculomegaly may develop only in late gestation or after birth, particularly with the X-linked hydrocephalus spectrum. The patients at risk for should be informed that a normal midtrimester sonogram does not rule out this condition. Couples with a previously affected child should receive genetic counseling, because sometimes a generic diagnosis of congenital hydrocephalus may hinder a more complex anomaly with significant genetic implications. For example, patients at risk for X-linked hydrocephalus spectrum should be offered DNA analysis, as the recurrence rate is high and midtrimester sonography is frequently unsuccessful.
Implications for sonographic screening: In all standard sonographic examinations, a view of the lateral ventricles should be obtained, and at least one of the atria should be visualized and assessed. A qualitative evaluation is acceptable, and the presence of the choroid plexus filling the cavity of the atrium, being closely apposed to both the medial and lateral wall of the ventricle, is indicative of normalcy. A quantitative approach is however favored, and a measurement less than 10 mm is considered normal between 15-40 weeks. Congenital ventriculomegaly may develop late in gestation, and a normal midtrimester exam does not exclude this condition.
Prognosis: In a recent review, isolated ventriculomegaly diagnosed in utero was associated with a postnatal survival rate of 70% and 59% of the survivors had normal developmental quotient at follow-up. These results may be biased however by the inclusion of cases with borderline enlargement of the ventricles. In one of the largest pediatric series, excluding cases with X-linked hydrocephalus and congenital toxoplasmosis, the survival rate was 62% at 10 years, and 50% of survivors had a low developmental quotient (< 60). Only 29% of infants attending school reached a normal academic level. Macrocrania at birth, ventricular size and age at surgery had no influence on the outcome. X-linked hydrocephalus spectrum carries a severe prognosis, being usually associated with severe neurological deficits and premature death.
Obstetrical management: A search for associated congenital anomalies, including fetal karyotyping) and a workup for congenital infections associated with hydrocephalus (i.e., toxoplasmosis, cytomegalovirus, rubella) is indicated. Before viability, the option of pregnancy termination should be offered to the parents. Little data exist to support any specific management plan in continuing pregnancies. There is no evidence that anticipation of delivery is beneficial. Most infants with ventriculomegaly do not have macrocrania, and therefore a trial of labor is indicated in vertex presentation. Cesarean section should be reserved for standard obstetrical indications. Whether cephalocentesis should be offered in cases with macrocrania to overcome cephalo-pelvic disproportion is debated. Cephalocentesis is indeed associated with a perinatal mortality in excess of 90 percent. Intrauterine treatment consisting of the implantation of a ventriculoamniotic shunt for the relief of intracranial pressure during gestation has been attempted. Although experience in animal models appears encouraging, the clinical application of these procedures remains undetermined. In a group of 39 treated fetuses, the perinatal mortality rate was 18 percent, and moderate to severe handicaps affected 66 percent of the survivors.
taken from here
Recovering from brain surgery
After any surgery, it is not unusual, at first, to feel worse than you did before. This can be depressing if you are not prepared for it. You have just had brain surgery. That is a lot for your body to cope with. The post-op swelling means it will be a while before you feel the benefit from having had your tumor removed.
After brain surgery, it is not uncommon to have dizzy spells or to get confused about where you are and what is happening to you from time to time. These episodes can come and go. They can be upsetting for your relatives and also for you. Your nurse and doctor will explain that this is normal and part of the recovery period.
The operation itself can often make your symptoms worse at first. Or you may notice symptoms that you didn’t have before. The swelling can cause
* Weakness
* Poor balance or lack of coordination
* Personality changes
* Speech problems
* Fits
This time can be particularly difficult for your loved ones. They may worry that your operation has not worked. But the symptoms will usually lessen and disappear as you recover. This may take only days. But it can take weeks or sometimes months.
Your surgeon will have given you some idea of what to expect in the way of recovery. For some people, recovery will be complete. You may be able to get back to the same fitness level you had before your tumour. And may be able to return to all your usual activities before long, including your job if you have one.
If you have long term problems
Because of the position of their tumour, some people have long-term problems with speech or with weakness of an arm or leg. This can take a long time to recover from. It may be hard for you to keep your spirits up through this time. But with effort and help from physiotherapists, speech therapists and other rehabilitation specialists, you will get a lot better.
Your rehabilitation will start as soon as you can get out of bed. You will gradually be able to do more and more for yourself. You may never quite recover to the same level of fitness as before your treatment. But your condition can and will improve to some extent. Your confidence will increase as you learn to manage with whatever level of disability you have to cope with. There is more about recovering after a brain tumour in the Living with a brain tumour section.
How Are Brain and Spinal Cord Tumors in Children Treated?
Children with central nervous system tumors may be treated by surgery, radiation therapy, and/or chemotherapy. Treatment is different for different kinds of tumors. Each child's treatment must be approached individually to give the child the best chance of cure. Long-term side effects of the treatment must also be considered. Because of this, children under 3 years old are usually not given radiation therapy. Instead, the treatment relies mainly on removing the tumor by surgery and chemotherapy.
This approach is based on the desire to delay the need for radiation therapy, which can cause developmental delay and problems with intellectual development. Even in older children, however, radiation treatment can cause problems. Radiation oncologists (doctors specializing in treatment of tumors with radiation) try very hard to limit the amount of normal brain tissue that they treat.The Key Statistics About Brain and Spinal Cord Cancers
Malignant brain and spinal cord tumors, the second most common cancers in children (after leukemia), account for about 17% of malignant tumors. Around 3,200 central nervous system tumors are diagnosed each year in children under the age of 20. About one fourth of these are considered benign tumors. The incidence (number per 100,000 children) of these cancers has not changed much in recent years.
Over one half of patients with childhood brain tumors (all types combined) survive longer than 5 years. The outlook varies according to the type and location of the tumor. For example, approximately 90% of astrocytomas of the cerebellum are cured by surgery.Brain and Spinal Cord Tumors in Children
Brain tumors are masses of abnormal cells that have grown out-of-control. In most other parts of the body, it is critically important to distinguish benign (noncancerous) tumors from malignant (cancerous) ones. Benign tumors are almost never life threatening. The main reason cancers are so dangerous is because they can spread throughout the body. Most brain cancers can spread through the brain tissue but rarely spread to other areas of the body. Even so-called “benign” tumors are can, as they grow, compress brain tissue, causing damage that is often disabling and sometimes fatal. For this reason, doctors usually speak of "brain tumors" rather than "brain cancers." The major distinction is how readily they spread through the rest of the brain central nervous system and whether they can be removed and not come back.
The central nervous system is the medical name for the brain and spinal cord. Central nervous system tumors of adults and children often form in different areas, develop from different cell types, and may have a different outlook and treatment. This document refers to children's tumors.
The brain is the center of thought, feeling, memory, speech, vision, hearing, movement, and much more. The spinal cord and special nerves in the head called cranial nerves carry messages between the brain and the rest of the body. These messages tell our muscles how to move, transmit information gathered by our senses, and help coordinate our internal organs. The brain is located within and protected by the skull. Likewise, the spinal cord is protected by the bones of the spinal column. The brain and spinal cord are surrounded and cushioned by a special fluid, called cerebrospinal fluid. Cerebrospinal fluid is produced by the choroid plexus, which is located in cavities within the brain called ventricles. The ventricles as well as the spaces around the brain and spinal cord are filled with cerebrospinal fluid.
Parts of the Brain and Spinal Cord
The brain and spinal cord are the 2 main parts of the central nervous system.
The main areas of the brain include the cerebral hemispheres, cerebellum, and brain stem. Each of these parts has a special purpose. Tumors of different parts of the central nervous system disrupt different functions and cause different symptoms. Any disease involving that particular location within the brain can cause these symptoms, and they do not necessarily mean a brain tumor is present. Also, tumors in different areas of the central nervous system may be treated differently and have a different prognosis (outlook for survival). In very young children, less than 3 years of age, itÂ’s often hard to tell which part of the brain is affected during its early development. Very young children may not have the usual symptomns coming from that part of the brain involved as would be seen in adults. In this age group the only symptoms may be nonspecific and include irritability, crying, poor feeding, or vomiting.
The 2 cerebral hemispheres control reasoning, thought, emotion, and language. They are also responsible for your planned muscle movements (throwing a ball, walking, chewing, etc.) and for taking in sensory information such as vision, hearing, smell, touch, and pain.
The symptoms caused by a tumor in a cerebral hemisphere depend on the part of the hemisphere in which the tumor arises. Common symptoms include:
* seizures
* trouble speaking
* a change of mood such as depression
* a change in personality
* numbness, weakness or paralysis of part of the body
* changes in vision, hearing, and sensation
The cerebellum controls coordination of movement. Tumors of the cerebellum cause difficulty with coordination in walking, difficulty with fine movements of arms and legs, and changes in rhythm of speech.
The brain stem contains bundles of very long nerve fibers (axons) that carry signals controlling muscles and sensation or feeling from the cerebrum to and from the rest the body. In addition, most cranial nerves (which carry signals to and from the face, eyes, tongue, and mouth) start in the brain stem. Special centers in the brain stem also control breathing and the beating of the heart.
Tumors in this critical area of the brain may cause weakness, stiff muscles, or problems with sensation, hearing, facial movement, and swallowing. Double vision is a common early symptom of brain stem tumors, as are problems with coordination in walking. Because tumors of the brain stem often intermingle with normal nerve cells and the brain stem is so essential for life, it may not be possible to surgically remove these tumors from the brain stem.
The spinal cord, like the brain stem, contains bundles of very long axons (wire-like extensions) that carry signals controlling muscles, sensation or feeling, and bladder and bowel control. Spinal cord tumors may cause weakness, paralysis, or numbness. Because the spinal cord is such a narrow structure, tumors arising within it usually cause symptoms involving both sides of the body (for example, weakness or numbness of both legs). This is different than tumors of the brain, which usually affect only one side of the body. Moreover, most tumors of the spinal cord arise below the neck after nerves to the arms have branched off the spinal cord, so that only lower body functions – bowel, bladder, or leg – are affected.
Tumors may also arise from cranial nerves. The most common cranial nerve tumor in children is optic glioma, a tumor of the optic nerve (the optic nerve is actually an extension of brain tissue to the eye) causing blindness. Tumors arising from other cranial nerves may cause hearing loss (acoustic nerve) in one or both ears, facial paralysis (facial nerve), or facial numbness or pain (trigeminal nerve). Tumors arising in the nerves of the peripheral nervous system (parts of the nervous system other than the brain and spinal cord) generally cause pain, weakness,and/or loss of sensation in the area served by that nerve. They can also weaken the muscles controlled by that nerve.
Types of Cells and Tissues in the Brain and Spinal Cord
The brain consists of different kinds of tissues and cells. Different types of tumors can start in these different cell and tissue types. These different types of tumors have varying outlooks for survival and may be treated differently.
Neurons: These are the most important cells within the brain. They send signals through the axons. Axons may be very short (in the brain) or 2 to 3 feet long (in the spinal cord). Electric signals carried by neurons determine thought, memory, emotion, speech, muscle movement, and just about everything else that the brain and spinal cord do. Unlike many other types of cells that can grow and divide to repair damage from injury or disease, neurons quit dividing about 1 year after birth (with a few exceptions). Neurons do not usually form tumors, but they are damaged by tumors that start nearby.
Glial cells: Most brain and spinal cord tumors develop from glial cells. There are 3 types of glial cells – astrocytes, oligodendrocytes, and ependymal cells. Tumors of glial cells are sometimes referred to as a group and called gliomas. A fourth cell type called microglia is part of the immune system and is not truly glial in origin. Normal glial cells grow and divide very slowly. Glial cells are the supporting cells of the brain and continue to increase in number until the child is 5 years of age. At this time, the brain reaches its maximum size and will be the same size throughout oneÂ’s lifetime.
* Astrocytes help support and nourish neurons. When the brain is injured, astrocytes form scar tissue that helps repair the damage.
* Oligodendrocytes make myelin, a substance that surrounds and insulates axons of the brain and spinal cord. This allows oligodendrocytes to help neurons transmit electric signals through axons.
* Ependymal cells line the ventricles within the central part of the brain and form part of the pathway through which cerebrospinal fluid travels.
* Microglia represent 10% to 20% of the total population of glial cells in the brain. They are the immune (infection fighting) cells of the central nervous system.
Neuroectodermal cells: These are primitive cells that are probably the remains of embryonic cells and are found throughout the brain. The most common tumor that comes from these cells is the medulloblastoma, which arises in the cerebellum.
Meninges: These are specialized tissues that line the cerebrospinal fluid-filled spaces surrounding the brain and spinal cord. The meninges help form the spaces through which cerebrospinal fluid travels.
Choroid plexus: The choroids plexus is the area of the brain within the ventricles that makes cerebrospinal fluid, which nourishes and protects the brain.
Pituitary gland and hypothalamus: The pituitary is a gland found at the base of the brain. The hypothalamus is a part of the brain next to the pituitary gland. Both of these tissues help regulate the activity of several other glands. For example, they control the production of thyroid hormone by the thyroid gland, the production and release of milk by the breasts, and the production of male or female hormones by the testicles or ovaries. They also produce growth hormone, which stimulates body growth, and vasopressin, which regulates water balance by the kidneys.
The growth of tumors in or near the pituitary or hypothalamus, as well as surgery and/or radiation therapy in this area, can interfere with these functions. Consequently, a child may have low levels of one or more hormones and may need hormone treatments to correct any hormone deficiency.
Pineal gland: The pineal gland is not strictly part of the brain. It is, in fact, an endocrine gland that sits between the cerebral hemispheres. Its function is probably to make melatonin, a hormone that responds to changes in light.
Blood-brain barrier: Unlike most other organs, there is a barrier between the blood and the tissues of the central nervous system (brain and spinal cord) that keeps many drugs from getting into the brain, including most chemotherapy drugs that are used to kill cancer cells. However, some chemotherapy drugs can cross the blood-brain barrier to treat some malignant brain tumors.
Types of Brain and Spinal Cord Tumors
Sometimes brain tumors are found not to have started in the brain but rather to have spread, or metastasized, from some other part of the body. Tumors that start in other organs and then spread to the brain are called metastatic brain tumors and those that start in the brain are called primary brain tumors. This is important because metastatic and primary brain tumors are usually treated differently.
In children, metastatic tumors to the brain are much less common than primary brain tumors. Unlike other cancerous tumors, tumors arising within the brain or spinal cord rarely metastasize to distant organs. They cause damage because they spread locally and destroy normal tissue where they arise. This document only covers primary brain tumors.
Gliomas: This is not a specific type of cancer. Glioma is a general category that includes glioblastoma multiforme, primitive neuroectodermal tumors, anaplastic astrocytoma, astrocytomas, oligodendrogliomas, ependymomas, brain stem gliomas and optic gliomas. Because this word is often used in discussing brain tumors, it is defined here in an attempt to reduce confusion with it.
Tumors can form in any type of tissue or cell within the brain or spinal cord. Some tumors contain a mixture of cell types. The most common brain and spinal cord tumors of children are astrocytomas. The second most common are primitive neuroectodermal tumors (23%), and the third most common are other kinds of gliomas such as brain stem gliomas (15%). Ependymomas are the fourth most common at 9%. All the others are fairly uncommon and account for only 3%.
Astrocytoma: Most tumors that arise within the brain itself start in brain cells called astrocytes, a kind of glial cell. These tumors are called astrocytomas. About half of all childhood brain tumors are astrocytomas. Many astrocytomas cannot be cured because they spread widely throughout, and intermingle with, the normal brain tissue. They are called infiltrating astrocytomas. Sometimes infiltrating astrocytomas spread along the cerebrospinal fluid pathways. But it is very rare for them to spread outside of the brain or spinal cord.
Infiltrating astrocytomas are classified as low grade, intermediate grade, or high grade. A pathologist (a doctor specializing in the diagnosis of diseases by laboratory tests) will grade them based on how the cells from a biopsy specimen (sample of the tumor) look under the microscope. Low-grade astrocytomas are the slowest growing and the most common type of astrocytoma in children. Intermediate-grade astrocytomas, or anaplastic astrocytomas, grow at a moderate rate. The highest-grade astrocytomas, glioblastomas, are the fastest growing.
There are some special types of astrocytomas that tend to have a particularly good prognosis. These are juvenile pilocytic astrocytomas and subependymal giant cell astrocytomas.
* Juvenile pilocytic astrocytomas most commonly occur in the cerebellum but also occur in the optic nerve, hypothalamus, brain, or other areas.
* Subependymal giant cell astrocytomas occur in the ventricles and are almost always associated with tuberous sclerosis (an inherited condition which may also cause epilepsy, mental retardation, and tumors of the skin and kidneys).
Certain tumors possibly of mixed glial and neuronal origin that occur in children and young adults and rarely in older adults also have a good prognosis. One such tumor is the pleomorphic xanthoastrocytoma and another is the dysembryoplastic neuroepithelial tumor. Although they appear malignant under the microscope, these tumors are relatively benign and most are cured by surgery alone.
Oligodendrogliomas: These tumors start in brain glial cells called oligodendrocytes. They spread or infiltrate in a manner similar to astrocytomas and, in most cases, cannot be completely removed by surgery. A small number of oligodendrogliomas, however, are associated with long-term survival of 30 or 40 years. Oligodendrogliomas may spread along the cerebrospinal fluid pathways but rarely spread outside the brain or spinal cord.
Optic Glioma: Optic gliomas are low-grade tumors of childhood and are frequently associated with an inherited condition called neurofibromatosis-type 1. These tumors, which arise from the optic nerve, can sometimes be treated successfully by surgery. At other times radiation therapy or chemotherapy may be required. The tumors are rarely lethal but may cause substantial visual loss.
Ganglioglioma: This tumor contains both mature neurons and glial cells. It has a high rate of cure by surgery alone or surgery combined with radiation therapy.
Primitive neuroectodermal tumors: Almost one fourth of brain tumors in children are of this type. They are rare in adults. When these arise in the cerebellum, they are called medulloblastomas. They are fast-growing tumors that can spread along the spinal cord and meninges but can be treated. Up to 50% of cases are cured by surgery and radiation therapy, sometimes with added chemotherapy. About 15% of childhood brain tumors are medulloblastomas.
Primitive neuroectodermal tumors are called pineoblastomas when they occur in the pineal gland. Other forms of primitive neuroectodermal tumors are all rapidly growing tumors that frequently spread throughout the cerebrospinal fluid pathways. The outlook for pineoblastomas is not as favorable as for medulloblastomas.
Ependymomas: Almost 10% of brain tumors in children are ependymomas. These tumors arise from the ependymal cells that line the ventricles or central canal of the spinal cord. Ependymomas may block the exit of cerebrospinal fluid from the ventricles causing the ventricle to become very large – a condition called hydrocephalus. Unlike astrocytomas and oligodendrogliomas, ependymomas usually do not spread or infiltrate into normal brain tissue. As a result, some but not all ependymomas can be removed and cured by surgery. Spinal cord ependymomas have the greatest chance of surgical cure. Ependymomas may spread along the cerebrospinal fluid pathways but do not spread outside the brain or spinal cord. Ependymomas represent about 9% of childhood brain tumors.
Choroid plexus tumors: These tumors arise in the choroid plexus within the ventricles of the brain. They are usually benign and cured by surgery (choroid plexus papillomas). However, they may also be malignant (choroid plexus carcinomas).
Craniopharyngioma: This type of tumor arises above the pituitary gland but below the brain itself. Most craniopharyngiomas are very close to the optic nerve, making surgical removal difficult, because of possible damage to the childÂ’s vision. They may also compress the pituitary gland and the hypothalamus causing hormonal problems. Some are cured by surgery; others require radiation therapy.
Schwannoma (neurilemoma): This type of tumor starts in Schwann cells that surround and insulate cranial nerves and other nerves. Schwannomas are usually benign tumors that often form near the cerebellum and in the cranial nerve, which is responsible for hearing and balance. They also arise from spinal nerves after they have left the spinal cord and can compress the spinal cord causing weakness, sensory loss, and bowel and bladder problems. These tumors are rare in children and when present in this age group, particularly if there is more than one, might suggest an inherited familial tumor syndrome such as neurofibromatosis.
Meningioma: This type of tumor arises from the meninges, the tissue that surrounds the outer part of the brain and spinal cord. Meningiomas cause symptoms by pressing on the brain or spinal cord. Meningiomas are much less common in children than in adults.
Meningiomas are benign and are usually cured by surgery. Some meningiomas, however, are located dangerously close to vital structures within the brain and cannot be cured by surgery. Meningiosarcomas are rare but very malignant (cancerous) tumors that may come back many times after surgery or, in rare occasions, spread to other parts of the body.
Chordoma: This tumor starts in the bone at the back of the skull or at the lower end of the spinal column. Chordomas may come back many times over a period of 10 to 20 years causing progressive neurologic damage and deterioration. But they usually do not spread or metastasize to other organs.
Germ cell tumors: Germ cell tumors develop from germ cells that normally form eggs in women and sperm in men. During normal embryonic and fetal development, germ cells migrate to the ovaries or testicles and develop into eggs or sperm cells. Sometimes, however, a few germ cells may not migrate properly and end up in abnormal locations such as the brain. They may then develop into germ cell tumors similar to those that can form in the ovaries or testicles.
Germ cell tumors of the nervous system usually occur in children, most often in the pineal gland or above the pituitary gland. The most common germ cell tumor of the nervous system is the germinoma, which can be cured by radiation therapy and possibly chemotherapy in almost all cases. Other tumors of germ cell origin such as choriocarcinoma or yolk sac tumors are rarely cured by surgery. Both radiation therapy and chemotherapy are used in their treatment and in some cases this may not control the tumor completely. Germ cell tumors can sometimes be diagnosed without a biopsy by measuring certain chemicals in the cerebrospinal fluid or blood.
Neuroblastoma: Another kind of nerve cell tumor, which is not a brain tumor, is called neuroblastoma. This is the third most common cancer in children. Neuroblastomas rarely develop in the brain or spinal cord; most develop from nerve cells inside the abdomen or chest. This type of cancer is most commonly diagnosed during early infancy. Neuroblastoma is discussed in a separate American Cancer Society document.
Brain Tumors that are not Gliomas
* Chordoma
* Craniopharyngioma
* Medulloblastoma
* Meningioma
* Pineal Tumors
* Pituitary Adenoma
* Primitive Neuroectodermal Tumors
* Schwannoma
* Vascular Tumors
Chordoma
Chordomas, which are more common in people in their 20s and 30s than in other age groups, develop from remnants of the flexible spine-like structure that forms and dissolves early in fetal development and is later replaced by the spinal cord. Although these tumors are often slow-growing, they can metastasize or recur after treatment. They are usually treated with a combination of surgery and radiation therapy.
Craniopharyngioma
Craniopharyngiomas occur in the region of the optic nerves and the hypothalamus, a structure near the pituitary gland. Like chordomas, they develop from cells left over from early fetal development. They produce problems with vision and hormonal problems, slowing the child's growth and causing poor regulation of water balance. The benign craniopharyngioma is a tumor that usually affects infants and children, although it sometimes occurs in adults. While only a decade ago it was considered inoperable and rarely curable by surgery, it can now be removed with minimal or no brain damage in many cases because of the precision afforded by the surgical microscope and microsurgical techniques. Occasionally, if this tumor continues to grow and cannot be removed surgically, radiation therapy is also necessary.
Medulloblastoma
Medulloblastomas are the most common primitive neuroectodermal tumor (PNET), representing more than 25% of all childhood brain tumors. They occur in children more often than in adults. Medulloblastoma most often arises in the cerebellum, located in the lower back part of the brain and causes symptoms that include headache, nausea, vomiting and problems with muscle coordination (ataxia). Unlike other primary brain tumors, medulloblastoma has a tendency to spread throughout the nervous system if it remains untreated. In unusual cases, medulloblastomas may spread outside the nervous system, to the lymph nodes, bone marrow, lungs or other parts of the body. In many cases, they are treated with surgery and radiation therapy alone. They are fast-growing tumors, but because they are very sensitive to radiation therapy and chemotherapy they can often be treated effectively.
Meningioma
A meningioma is a common brain tumor that originates from the meninges, the thin membranes or lining that cover the brain and spinal cord. As they grow, meningiomas compress adjacent brain tissue. Meningioma symptoms are often related to this compression of brain tissue, which can also affect cranial nerves and blood vessels. In some cases, meningioma growth can also extend into the bones of the head and face, which may produce visible changes. Meningiomas account for about 27% of all primary brain tumors and tend to affect more women than men. Most meningiomas are considered benign tumors. However, unlike benign tumors elsewhere in the body, benign brain tumors can
sometimes cause disability and may sometimes be life threatening. In many cases meningiomas appear to grow slowly. Other meningiomas grow more rapidly or have sudden growth spurts. There is no way to predict the rate of growth of a meningioma or to know for certain how long a specific tumor was growing before diagnosis. Although most people develop a single meningioma, it is
also possible to have several tumors growing simultaneously in different parts of the brain and spinal cord. Because recurrent tumors cannot be predicted, it is very important for meningioma survivors to receive regular follow up scans as part of their lifetime health care in order to avoid critical care being neglected.
Pineal Tumors
These tumors arise in the region of the pineal gland, a small structure deep within the brain. They account for about 1% of brain tumors, but make up 3% to 8% of the intracranial tumors that occur in children. At least 17 different types of tumors may occur in this area, many of which are benign. The three most common types of pineal region tumors are gliomas, germ cell tumors and pineal cell tumors. Surgery is absolutely necessary to obtain a sample of tumor tissue so the pathologist can confirm a precise histological diagnosis, which is essential in planning the appropriate therapy. Benign pineal tumors can be removed surgically. The germinoma, the most common malignant tumor in this area, can be cured in more than 90% of patients. Other malignant germ cell tumors occurring in this region are treated with chemotherapy followed by radiation therapy. Over the past 5 years, the prognosis for children with pineal tumors has improved dramatically.
Pituitary Adenoma
The pituitary gland is a small oval structure located at the base of the brain in the center of the head, behind the eyes and optic nerve. It is about the size of a pea but is very important because it secretes several chemical messengers known as hormones, which help control the body's other glands and regulate growth, metabolism, maturation and other essential body processes. A tiny tumor located just next to the gland, pituitary adenomas account for about 10% of brain tumors. Doctors classify pituitary tumors into two groups - secreting and nonsecreting. Secreting tumors release unusually high levels of pituitary hormones, triggering a constellation of symptoms. They are usually much smaller than the gland when they begin to cause symptoms and the symptoms depend on the tumor's size and the kind of hormone the tumor secretes. Prolactin-secreting adenomas affect sexual characteristics and cause impotence in men. Adenomas secreting growth hormone cause acromegaly (abnormal body growth, enlarged facial features, hands and feet) and gigantism (excessive size and stature). The less common adrenocorticotropic hormone-secreting adenoma causes Cushing's disease. Some adenomas secrete a combination of these or other hormones and some secrete none. Almost all adenomas are benign, but their slow expansion compresses normal structures that surround it, suppressing normal pituitary function and sometimes causing headaches or problems with vision. Pituitary adenomas rarely metastasize or spread to other areas of the body. They are removed in an operation using microsurgical techniques, a very successful form of treatment for the majority of patients.
Primitive Neuroectodermal Tumors
Primitive Neuroectodermal Tumors (PNETs) usually affect children and young adults. Their name reflects the belief, held by many scientists, that these tumors spring from primitive cells left over from early development of the nervous system. PNETs are usually very malignant, growing rapidly and spreading easily within the brain and spinal cord. In rare cases, they cause cancer outside the central nervous system. Medulloblastomas are the most common PNET. Other more rare PNETs include neuroblastomas, pineoblastomas, medulloepitheliomas, ependymoblastomas and polar spongioblastomas. Because their malignant cells often spread in a scattered, patchy pattern, PNETs are difficult to remove totally through surgery. Doctors usually remove as much tumor as possible with surgery then prescribe high doses of radiation and, in some cases, chemotherapy.
Schwannoma
Schwannomas arise from the cells that form a protective sheath around the body's nerve fibers. They are usually benign and are surgically removed when possible. One of the more common forms of schwannoma affects the eighth cranial nerve, which contains nerve cells important for balance and hearing. Also known as vestibular schwannomas or acoustic neuromas, these tumors may grow on one or both sides of the brain.
Vascular Tumors
These rare, noncancerous tumors arise from the blood vessels of the brain and spinal cord. The most common vascular tumor is the hemangioblastoma, which is linked in a small number of people to a genetic disorder called Von Hippel-Lindau disease. Hemangioblastomas do not usually spread and doctors typically treat them with surgery.
(This information was taken respectively from here)
Shunt for Hydrocephalus (Water in the Brain)
Taken from here
The most common surgery for the treatment of hydrocephalus (water on the brain), is the insertion of a shunt - a device that diverts fluid from the brain into the abdominal cavity where it is safely absorbed into the blood stream. Though a shunt may be inserted in infants, children and adults, the procedure is essentially the same regardless of the size of the patient.
Anatomy and Physiology
- In a normal brain there are four fluid filled spaces called ventricles (Figure 1)
- There are two large ventricles (lateral ventricles) located on either side of the brain
- Two other ventricles (the third and fourth ventricles) are placed along the midline
- There are two large ventricles (lateral ventricles) located on either side of the brain
- Within the ventricles are tufts of vascular tissue called the choroid plexus. As blood flows through the choroid plexus it distills a clear watery fluid into the ventricles called cerebrospinal fluid, or CSF
- CSF flows from the ventricles towards the surface of the brain and returns to the blood (Figure 2)
- From the lateral ventricles the CSF flows into the centrally placed third ventricle and then through a narrow channel called the aqueduct, into the fourth ventricle
- CSF then flows over the surface of the brain in the subarachnoid space, an area between the brain and the membrane (dura) lining the inside of the skull
- The CSF then returns to the blood by passing into the large veins that drain the brain called sinuses

- From the lateral ventricles the CSF flows into the centrally placed third ventricle and then through a narrow channel called the aqueduct, into the fourth ventricle

Picture taken from www.Yoursurgery.com
Craniotomy
A craniotomy is the surgical removal of a section of bone (bone flap) from the skull for the purpose of operating on the underlying tissues, usually the brain. The bone flap is replaced at the end of the procedure. If the bone flap is not replaced, the procedure is called a craniectomy. A craniotomy is used for many different procedures within the head, for trauma, tumor, infection, aneurysm, etc.
Anatomy
- At birth the bones that make up the cranium or skull are separated, allowing the head to pass through the birth canal. (Figure 1)
- As the individual matures, the bones fuse together so that by late teens the bones form a solid union
- The various bones of the skull are the frontal, parietal, temporal, occipital, and sphenoid, (Figure 2)
- The scalp covers the skull
- Within the skull lie:
- The brain, which is divided into four major parts- the right and left cerebral hemispheres, the cerebellum and the brainstem (Figure 3).
- The brain, which is divided into four major parts- the right and left cerebral hemispheres, the cerebellum and the brainstem (Figure 3).
- The cerebral hemispheres form the largest portion of the brain and can be regarded as the 'thinking' part of the brain and are involved in movement, sensations, speech and creation of ideas
- Each cerebral hemisphere is divided into four lobes - frontal, parietal, temporal and occipital
- The surface of the hemispheres is folded upon itself and presents as various grooves (sulci) and bulges (gyri). The two cerebral hemispheres are connected across the midline by a large band of brain fibers called the corpus callosum that transmit nerve impulses between the hemispheres
- The cerebellum lies at the back of the brain under the occipital bone and is involved in fine tuning movement
- The brainstem lies in front of the cerebellum and is attached above to the cerebral hemispheres, behind to the cerebellum and below to the spinal cord
- The meninges (the membranes that line the inside of the skull (dura) and cover the brain (pia-arachnoid). A large fold of dura called the falx lies above the corpus callosum and separates the cerebral hemispheres. (Figure 4) Another large fold of dura, the tentorium, separates the cerebral hemispheres from the cerebellum. The brainstem passes through a hole in the front of the tentorium. The space that lies beneath the tentorium, which contains the cerebellum and brainstem, is called the posterior fossa
- The blood vessels that feed the brain
- The cerebrospinal fluid (the fluid that bathes the brain) originates within the ventricles (spaces) within the brain
Pituitiary Tumor
by John R. Mangiardi, M.D. and Howard Kane, Wm.
The pituitary gland is a half-breed in many ways. It is not really a part of the brain, but rather hangs beneath it. Half of the gland comes down from the brain (the posterior lobe which controls the body's water levels and secretes the hormone ADH -- anti-diuretic hormone). The other half comes from tissues originating from the roof of the embryonic mouth, the anterior lobe which controls sex hormone levels, lactation, growth hormone, body steroids, and the thyroid gland).
The pituitary is responsible for almost all of the body's hormonal systems, taking all of its cues from the hypothalmus, the hidden and very deeply located Grand Wizard of the brain. The hypothalmus also controls such activities as body temperature, sexual drive, appetite, blood glucose levels, and sleep/arousal behavior patterns.
As elsewhere in the brain, tumors of the pituitary gland behave according to their cell of origin. Most of these tumor are truly benign, although on occasion they may prove to be malignant (pituitary carcinomas). The list of cells of the pituitary determines the tumor types, as well as the clinical syndromes related to each. Almost all have the good prognosis which calls for total removal. On the other hand, almost all can eventually become "malignant by position."
This is especially true when the tumors grow off to either side, involving the jam packed structures behind both eyeballs, called the "cavernous sinuses." The pituitary gland is located exactly between these two structures, which contain the nerves that control eye movement and the major arteries that feed the brain (carotid arteries, the veins that drain the eyes and other nerve related structures). Therefore, these tumors may occasionally present the patient with double vision, or even something called "Pituitary Apoplexy" (severe sudden headache, loss of and/or double vision, protruding eyeball).
Each tumor, because of its extraordinarily high hormonal output, creates a characteristic clinical syndrome that brings attention to the tumor. Because the pituitary gland is located directly beneath the place where the nerves cross, coming from the eyes to the brain (the optic chiasm), many tumors also present -- along with the hormonal problems listed below -- loss of peripheral vision.
Hormonal Problems
Gigantism: This syndrome is caused by pituitary tumors on the growth hormone secreting cells of the pituitary gland. Remember Lurch from the James Bond movie -- large hands, protruding jaw, severe arthritis, huge size, protruding eyebrows, plus other systemic problems -- a classic example of gigantism.Cushings Disease: This syndrome is caused by tumors on the ACTH (Adrenal Corticotrophin Hormone) secreting cells of the pituitary gland. Patients with this problem develop fat deposits in strange places (Moon face, Buffalo hump on the back of the neck), spontaneous scarring of the skin along the belly that look striated, pimples in adults, high blood pressure and elevated body temperature. These tumors are usually so small that the surgeon might have a difficult time finding the little "bad pearl" in the gland during surgery. This is the one time when small can be bad, especially if the surgeon is unable to locate and remove the tumor! ACTH secreting tumors, although small and troublesome, are readily cured by surgery alone.
Prolactin Syndrome: This syndrome is caused by tumors on the prolactin secreting cells of the pituitary gland. The tumors are the most common of all the pituitary tumors. Production of breast milk in women who are not pregnant, loss of menstrual cycle, and loss of bone calcium are all hallmarks of this tumor. When small, it may be cured; when large, it may cause visual problems and require other (e.g. radiation) therapy. Many women with this tumor visit their gynecologist thinking that they might be pregnant.
Growth hormone secreting tumors may very occasionally be treated with drugs, but most often must be removed surgically.
Non-Secreting Tumors: Can be treated by surgery and/or radiation. These patients almost always have problems with vision, as the hormonally quiet tumor grows to oversized proportions, actually growing to the point of lifting up and stretching the optic nerves (especially where the nerves from both eyes cross as they travel to the brain). The treatment of these tumors is variable. Prolactinomas are most often treated non-surgically with drugs that inhibit prolactin production (parlodil). Microprolactinomas sometimes never really grow over long periods of time, and do not require surgery.
All pituitary tumors can be treated by radiation, especially with the improvements brought on by focused beam radiation (liner accelerator and proton beam). The idea of radiosurgery originated in this venue. One serious problem with radiation has been loss of function in the remainder of the pituitary gland, requiring patients to depend on hormone supplements for the continuation of their lives. Another has been the inability to quickly reverse visual loss in large tumors using radiation. On the other hand, radiation has been used as a very successful adjunct in larger tumors that pose a threat to long term survival.
Surgery on Pituitary Tumors
The wonderful surgical achievement of the modern age is the combination of improved lighting, the surgical microscope, and computer assisted navigational instrumentation now used in the O.R. The pituitary gland lies just above the air spaces in the nose (if you'd stick a pencil through your nose hard enough, you'd end up in your pituitary gland). In fact, the word "pituita" (like the word "ptuey!") refers to the not so delicate production of "snot." In medieval times in Europe, and in China today, it is thought that "pituita" was something good to get rid of, serving as a relief valve for bad humors of the brain. In other words, spitting was good for the soul as well as one's health.Thus, by traversing the structures just beneath the skull through the nasal cavities, brain surgery can be avoided and the risk of approaching the pituitary gland can be enormously reduced.
Decision Making for Pituitary Tumor Surgery
It's an Emergency: Patients who have a large pituitary tumor (and often don't even know about it) will occasionally develop a kind of pituitary "stroke," called pituitary apoplexy. This occurs after the tumor outgrows its blood supply and suddenly enlarges (due to swelling) after it infarcts (a type of local stroke), or begins bleeding within the tumor. The enlargement causes severe headache and/or double vision, because the nerves that control the eye located next to the gland are pressed upon. It can also cause loss of vision (because the gland swells upward, pressing from beneath the optic nerves above). Surgical decompression is an emergency procedure because permanent blindness may result if left untreated.Surgery Is the Best Way to Go
Cushing's Disease: Because the tumor is so small, a cure is possible when removed. Thus, surgery is the best way to go. Moreover, the remainder of the gland is left intact, and will function normally thereafter.Large tumors with liquified (necrotic) centers: In these cases, the surgery is easy, and the improvement is immediate. Any remaining tumor beyond the confines of the surgical field can be safely treated by other standard therapies (e.g. radiation therapy).
Medium sized tumors: Still within the confines of the pituitary gland. As with the Cushing's tumors, the tumor can be completely removed and the gland saved for normal function.
A 'Hold' on the Surgery
Prolactinomas: Should always be treated initially with medication (anti-DOPA agents such as Parlodil, Bromocriptin, etc.). Even large tumors that most need to be treated by either surgey or radiosurgery should be pretreated with these drugs to shrink the tumor away from vital brain structures before surgery should be contemplated.Microprolactinomas: Surgically, nothing need be done for as long as possible. Some of these tumors appear to just sit there for years, even decades.
Extremely large tumors that cause little visual or other brain problems: In treating these tumors, particularly in older people, the physician should consider radiation therapy to stop the progression of these tumors. A surgical cure is usually not an option, for the substantial surgical risk entailed.
Pineal Region Tumor
As mentioned in the PRIMER, the pineal gland hangs on the brain behind its very center. French 18th century scientist Rene Descartes thought that the pineal gland was the core of the soul, noting that it was the only unpaired organ in the body, and located in the center of the brain. He thought that the gland controlled the movement of the various body "humors." In fact, the pineal gland is the "third eye" of the brain, and is responsible for telling the brain when it is day or night. It also controls the body's hormonal systems, sleep-wake cycle, and other so-called "circadian" body rhythms. It is in essence, the body's internal clock."
The gland produces a hormone called melatonin, (now a popular over-the-counter seller). Melatonin levels are what directly influence the function of various brain centers (appetite, sleep, the hypothalamus and pituitary gland). The cells of the pineal gland are unique in that they are not related to either the support cells of the brain (i.e. astrocytes, etc.), nor to the brain neurons. Rather, they develop on their own. For this reason, true pineal cell tumors are quite different from other brain tumors.
The pineal gland is also filled with brain support cells (astrocytes) and has a very dense input of nerve fibres coming from the eyes via a very circuitous and complicated route. Thus, the tumors of the astrocyte support cells of the pineal region are the same as the astrocytomas and GBM.
Pineal tumors, themselves, vary. They include:
- Pinealocytoma ("benign" pineal cell tumor)
- Pineoblastoma (more aggressive pineal cell tumor)
- Pineal germinoma ( aggressive primitive cell tumor growing in the pineal region)
- Pineal teratomas (rare tumors of multiple cell types that grow in the pineal region)
- Pineal Cysts (most often not treated, unless large enough to cause hydrocephalus or visual symptoms)
A combination of CAT scan, MRI, and spinal fluid studies (including "markers" such as AFP, and spinal fluid cytology), will aid the surgeon to deliver a very good guess or outright diagnosis.
Treatment Decisions -- Tumor Biopsy
With all of these possibilities in such a small area, it is no wonder that most physicians feel strongly that at least a biopsy should be taken prior to considering a treatment course.
- Surgical Removal. These tumors should be approached surgically at first, with a best effort approach to remove as much as, if not all, of the tumor. With current surgical technology, the down side has essentially been reduced to well below the 2% risk mark, making the decision to operate much less difficult than previously. Today, the surgery can be minimized so that recovery time is shortened. Some patients with larger tumors may also develop hydrocephalus, requiring the placement of a shunt at the time of surgery or thereafter.
- Radiation. Some centers perform radiation, at least in a "test" dose (in the case of the germinoma) since some of the primitive tumors are quite sensitive, a full course of radiation might be considered.
- Craniospinal axis. If the spinal fluid cytology is positive, so-called "craniospinal" axis therapy may be considered. This is done because some of these tumors will "seed" down from the brain into the spinal canal.
- Chemotherapy. Many chemotherapy choices may be available. Some of these tumors are quite sensitive to chemotherapy, accounting for the vigorous chemo-approach of many neurooncologists to such lesions.
Meningioma
by John R. Mangiardi, M.D. and Howard Kane, Wm.
The meningioma is the neurosurgeon's "friend" and often his most enduring challenge. For both the physician and patient, this tumor carries a true tag of benign. It also carries the possibility of "cure" in approximately 80% of cases. Thus, the long-term outcome for a patient with this tumor is a direct function of the skill and assiduousness of the surgeon who removes it.
Elsewhere in the Brain Surgery Information Center's Primer on Brain Tumor Biology, it was mentioned that "benign" often does not really mean benign. Be assured that in this case, the tumor really is benign.
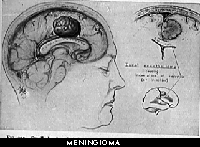
As mentioned earlier in the Primer, each type of brain tumor arises from a specific cell type. The cell of origin for the meningioma is call the arachnoid cap cell, found on the surface coverings (called meninges) of the brain in the paccionian granulations. These serve as the one-way valve system between the water system of the brain and the veins that drain from the brain to the heart.
Interestingly, these tumors have an embryologic relationship with cells found in the muscle layer of the utereus. In fact, it is exceedingly difficult for the pathologist to distinguish the meningioma from the fibroid tumors of the utereus under the microscope. Also, they share the characteristic female hormonal receptors (estrogen and progesterone) on their cell surfaces. This characteristic has lead to the testing of anti-estrogen receptor agents, such as tamoxifin, as a growth-inhibiting agent in these tumors. Clinical studies to date have failed to provide siginificantly positive results.
Meningiomas are rarely malignant in their behavior. But when malignant, meningiomas grow rapidly and are destructive; they are quite difficult to treat, and recur oftentimes in less than a year after surgical removal. They are also difficult for the pathologist to diagnose under the microscope. Probably the only finding that correlates well with the diagnosis is that of numerous cells seen in division ("mitosis"). The pathologist may occasionally speak of brain and skull invasion, cells with an abnormal appearance, or other bizarre findings, however none of these completey fit the diagnosis. Ultimately, the diagnosis is determined by the activity of the particular tumor over time.
A cousin to the meningioma is the hemangiopericytoma. The cell of origin for this tumor is the perivascular pericyte (located around blood vessels). Although very similar to the benign meninigiomas, these tumors tend to recur with great rapidity (less than one year) and frequency. Some physicians classify these tumors with the malignant meningiomas.